We just established different patient-derived fibroblasts as models for various lysosomal storage diseases (LSDs), offering a valuable tool for drug screening.
LSDs are a group of rare genetic disorders, characterized by deficiencies in specific enzymes that usually facilitate the lysosomal degradation process, leading to the accumulation of undegraded substances within the lysosome. These diseases can lead to significant impairments in physical and cognitive functions, causing developmental delays, organ dysfunction, and a shortened lifespan. Some LSDs may manifest already in infancy or childhood, leading to rapid deterioration of health.
We now started to characterize fibroblasts from Gaucher, Pompe and Niemann-Pick disease patients by analyzing pathological differences between patients-derived cells and healthy controls. Enzyme activity via 4MUG assay as well as changes in lysosome morphology and size, analyzed as total Lysotracker signal, was evaluated as an increased Lysotracker signal indicates an increased lysosomal size due to changes in morphology.
Assessment of β-glucocerebrosidase (GCase) activity in fibroblasts from Gaucher disease patients and α-glucosidase (GAA) activity in fibroblasts from Pompe disease patients in comparison to healthy controls revealed pronounced impairments in enzyme activity of diseased fibroblasts. Lysotracker imaging of fibroblasts from Gaucher, Pompe, and Niemann-Pick (NPC) patients further highlighted significant differences in lysosomal morphology compared to healthy cells.
Gaucher patient-derived fibroblasts exhibited strongly reduced GCase activity, supported by Lysotracker staining, showing an increased signal compared to healthy cells, reflecting enlarged lysosomes when compared to a healthy control (Figure 1).
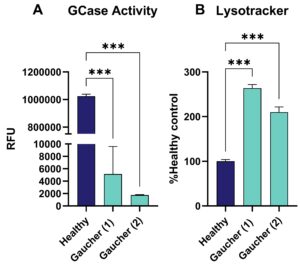
Figure 1: Gaucher Disease: (A) β-glucocerebrosidase (GCase) activity of Gaucher patient-derived and healthy individual-derived fibroblasts assessed via 4MUG assay given as relative flourescent unit (RFU) as well as (B) Lysotracker signal displayed as percent of healthy control. One-way ANOVA followed by Dunnett’s multiple comparison test. ***p<0.001.
Similarly, Pompe patient-derived cells displayed heavily impaired GAA activity, with Lysotracker staining revealing a significantly increased signal compared to healthy cells (Figure 2).
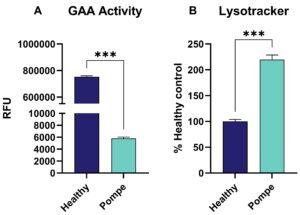
Figure 2: Pompe Disease: (A) α-glucosidase (GAA) activity of Pompe patient-derived and healthy individual-derived fibroblasts assessed via 4MUG assay and given as relative flourescent unit (RFU) as well as (B) Lysotracker signal displayed as percent of healthy control. Unpaired t-test. ***p<0.001.
Two distinct fibroblast lines derived from Niemann-Pick disease (NP) patients exhibited a significantly higher number of lysosomes compared to healthy controls after 12 hours of Lysotracker incubation (Figure 3).
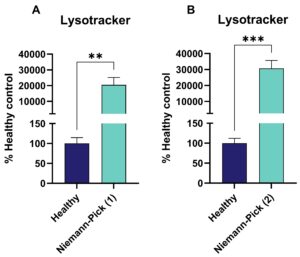
Figure 3 Niemann-Pick Disease: Lysotracker signal of two different Niemann-Pick type C patient-derived fibroblasts displayed as percent of healthy control. Unpaired t-test. **p<0.01; ***p<0.001.
These findings underscore the potential of LSD patient-derived fibroblasts as a robust tool for drug testing as they realistically mimic disease characteristics. These cell lines can be used as complementary tool to the respective mouse model available, including 4L/PS-NA and GBA D409V KI mice, NPC1-/- mice, and 6neo mice to model Gaucher disease, Niemann-Pick type C1 disease, and Pompe disease, respectively.
Our ongoing efforts include extensive characterizations of various LSD fibroblast lines, comprising of other LSDs such as Krabbe and Fabry diseases. This research contributes to advancing our understanding of LSDs and holds promise for the development of targeted therapeutic interventions.
Contact us today to get your study in patient-derived fibroblasts started!








